Epoxide Reactions: Acid-Assisted Ring Opening (H₃O⁺, ROH/H⁺, HX)
Brønsted acids such as H₃O⁺ protonate epoxides, polarising the C–O bonds so the more substituted carbon carries greater positive character. Weak or neutral nucleophiles (H₂O, ROH, RCO₂H) now attack from the backside, giving anti products with inversion at the attacked centre. Stronger nucleophiles such as halides in HX follow the same anti pathway; they usually favour the less hindered carbon unless a tertiary or benzylic site offers superior stabilisation. The figures below use 2,2-dimethyloxirane under H₃O⁺ to spotlight the Markovnikov outcome.
Quick Summary
- Reagents/conditions: H₃O⁺ (dilute mineral acids), ROH/H⁺ (alcoholysis), or HX (HCl/HBr/HI; often anhydrous).
- Regioselectivity: Attack at the more substituted carbon (benzylic/tertiary > secondary > primary) for H₂O/ROH/RCO₂H. Halides in HX default to the less hindered carbon unless a tertiary/benzylic site stabilizes positive charge.
- Stereochemistry: Backside attack on the protonated epoxide gives inversion at the attacked carbon and overall anti (trans) 1,2-addition.
- Products: trans-1,2-diols (H₂O), trans-alkoxy alcohols (ROH), trans-halohydrins (HX), or trans-acyloxy alcohols (RCO₂H).
- No rearrangements: The opening is concerted (SN1/SN2 hybrid); discrete carbocations and 1,2-shifts are rare under standard acidic conditions.
Mechanism — Acid-Assisted Epoxide Opening (4 Frames; arrows A–H)




Mechanistic Checklist (Exam Focus)
- Protonate first; the oxonium directs the site of attack.
- H₂O / ROH / RCO₂H attack the more substituted carbon (benzylic/tertiary > secondary > primary).
- HX: halide attack is still backside/anti. Less substituted carbons react fastest unless a tertiary/benzylic position stabilises positive charge.
- Anti stereochemistry dominates: inversion at the attacked carbon gives trans products.
- Ring strain drives the opening; larger cyclic ethers do not react nearly as readily under comparable conditions.
Worked Examples
2,2-Dimethyloxirane + H₃O⁺ → Markovnikov trans diol

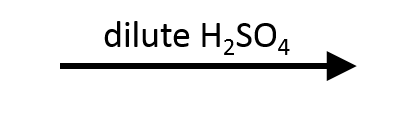

Protonation by H₃O⁺ makes the tertiary carbon most electrophilic, so H₂O adds there. Deprotonation furnishes the anti diol with the tertiary alcohol on the former epoxide carbon.
2,2-Dimethyloxirane + HBr → trans tert-bromohydrin

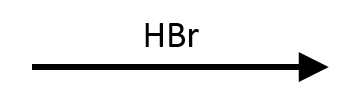

Even though halides often choose the less substituted carbon, the tertiary site here wins because the protonated ring can stabilise the developing cation. The result is a trans bromohydrin with Br at the tertiary carbon.
2,2-Dimethyloxirane + MeOH/H⁺ → trans tert-methoxy alcohol

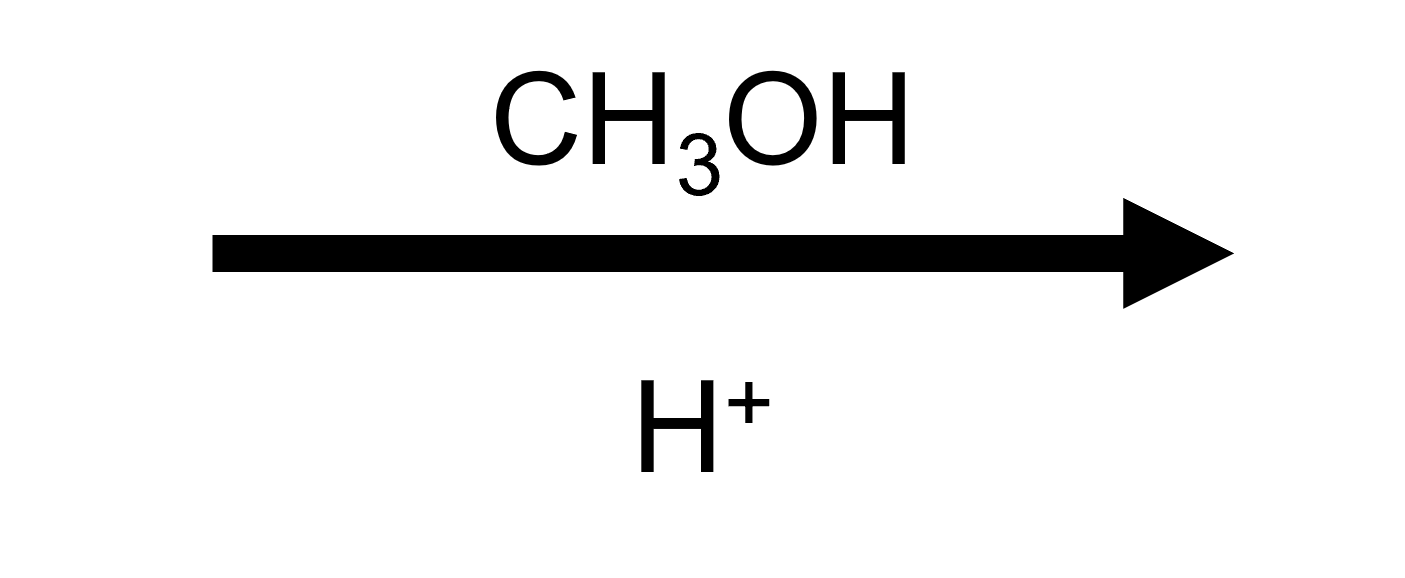

Methanol attacks just like water but leaves behind a protonated OR group. Acid-base work-up removes that proton to reveal the anti methoxy/HO pairing.
2,2-Dimethyloxirane + EtOH/H⁺ → trans tert-ethoxy alcohol

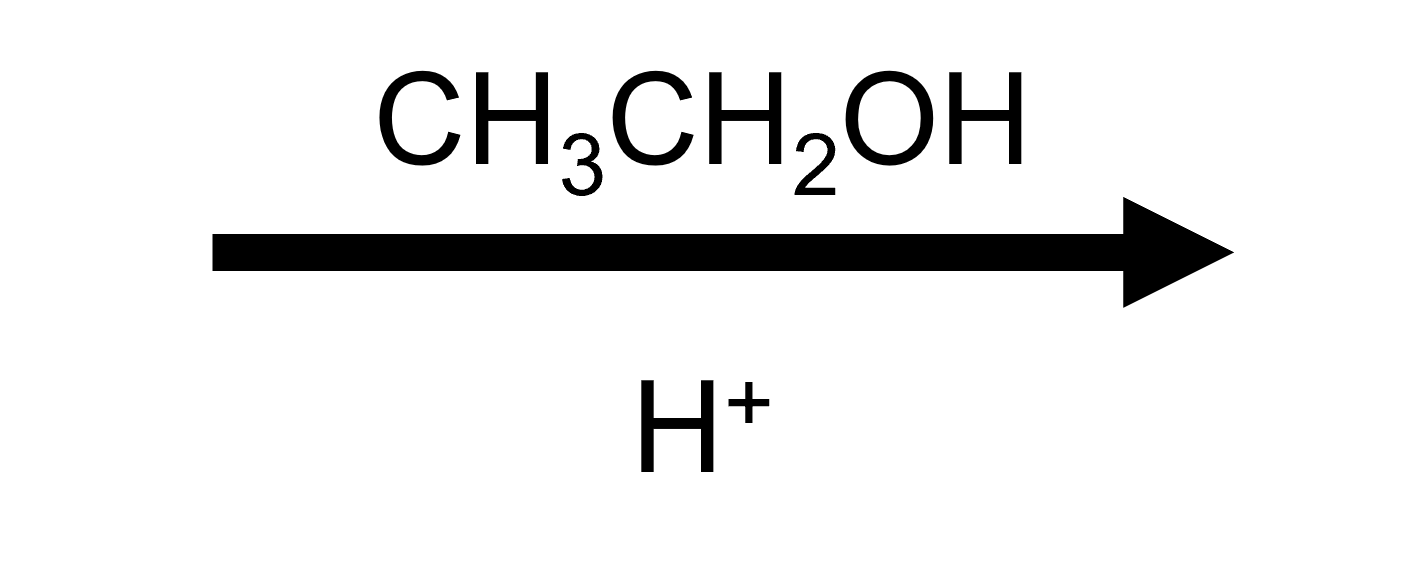

Ethanol follows the same script as methanol, delivering a trans ethoxy/HO pair after deprotonation. Compare the two to emphasise how changing ROH swaps the alkoxy group in the product.
Scope & Limitations
- Favourable substrates: Aryl, benzylic, allylic, and simple alkyl epoxides. Tertiary centres strongly direct attack to themselves under acidic conditions.
- Regiochemical rules: Weak/neutral nucleophiles attack the more substituted carbon; halides favour less substituted carbons unless tertiary/benzylic positions are available.
- Cyclic epoxides: Opening gives trans-1,2-disubstituted products; chair conformations dictate axial/equatorial placement.
- Acid-sensitive groups: Protect or avoid strongly acid-labile functionality (acetals, some protecting groups) when using Brønsted acids.
- HF variants: Fluorohydrin synthesis often requires specialized HF complexes (HF·pyridine); adjust conditions accordingly.
- Amines: Protonated amines are poor nucleophiles under these conditions—use basic openings for azide/amine capture instead.
Practical Tips
- Use dilute strong acids (0.1–1 M H₂SO₄, HClO₄, TsOH) with ample H₂O/ROH to minimize rearrangements or polymerization.
- For halohydrins, employ anhydrous HX and keep the medium as dry as practical so the halide remains a strong nucleophile.
- In cyclic systems, draw chair conformations to show anti attack from the less hindered face.
- Quench strongly acidic mixtures cautiously and neutralize residual acid before workup. Capture halogen waste appropriately.
Exam-Style Summary
Epoxide activation by protonation polarizes the ring, pushing positive character onto the more substituted carbon. Backside nucleophilic attack opens the ring with inversion to give overall anti products. H₂O/ROH/RCO₂H attack the more substituted carbon; halides usually attack the less hindered carbon unless a tertiary/benzylic site is present. Products are trans diols, alkoxy alcohols, or halohydrins—no rearrangements required.
Interactive Toolbox
- Mechanism Solver — Use Mechanism Solver to see each step of the acid-assisted epoxide opening mechanism along with descriptions of each step!
- Reaction Solver — Quickly find the product of any epoxide reacted with H₃O⁺, ROH/H⁺, or HX!
- IUPAC Namer — Learn the naming ins and outs of epoxide starting materials and their trans addition products.
Related Reading
- Prefer heavy-metal oxidants? Compare with Epoxide Formation via Halohydrin Cyclization for the complementary base-induced pathway.